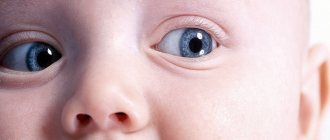
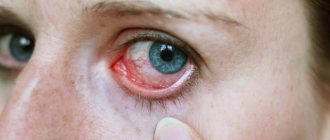
Retinal cancer usually affects young children under five years of age. In adults, this pathology is practically not detected. This disease is called retinoblastoma - a malignant formation that forms in children on the retina of the eye, and also affects its tissues and orbit. Anyone can experience the appearance of a cancerous tumor, regardless of age and gender, and cancer can affect a variety of human organs, even the eyes.
Retinoblastoma is found equally often in children of different sexes. The peak of the disease is between 2 and 3 years. Of all cases of cancer diagnosed in children, about 5% are retinoblastoma. If the pathology is not recognized in a timely manner, it can not only cause vision loss, but also, as it progresses, begin to spread metastases.
Pathogenesis
The development of a neoplasm in the retina can be unilateral or bilateral. In the latter case, in most cases we are talking about the hereditary nature of the pathology. Bilateral retinal cancer is diagnosed in a quarter of patients.
Unilateral retinoblastomas are often not inherited, but even in this case there is a clear connection with chromosomal aberrations. Typically, a deletion occurs in the long arm of the 13th pair of chromosomes (the RB1 gene is damaged). With such damage, patients not only develop retinoblastoma, but are also at risk of developing osteosarcoma.
Scientists believe that retinoblastoma is formed from cells of the neuroectoderm of the retina. Tumor cells can be at different stages of differentiation. With large tumors, cavities of decay, necrosis, and calcifications are formed. Retinoblastoma itself consists of small undifferentiated cells that have a large nucleus. This type of tumor is characterized by the presence of multiple growth foci in the retina. In most cases, patients die from tumor metastasis to the bones, central nervous system, bone marrow and its growth along the optic nerve.
Diagnostics
When a child experiences symptoms indicating retinal cancer, an urgent consultation with an ophthalmologist is necessary. Oncologists and neurologists also take part in the diagnosis of retinoblastoma. First of all, the doctor conducts an external examination and checks vision. The presence of retinoblastoma may be indicated by strabismus, a weak reaction to light and dilation, as well as leukocoria.
If a child has these signs of the disease, additional tests are prescribed to make an accurate diagnosis:
- ophthalmoscopy,
- fluorescein angiography,
- radiography,
- computed tomography (CT),
- magnetic resonance imaging (MRI),
- ultrasound examination (ultrasound),
- biopsy.
Carrying out tomographic studies is necessary to assess the extent of the tumor. An intraocular biopsy is prescribed to a child only if there is an urgent need for this study, since this procedure may lead to the dissemination of malignant cells in the eye. Only after receiving the results of all the studies performed, the doctor can prescribe a further course of therapy.
Histogenesis
It was not possible to fully determine the histogenesis of retinoblastoma. The famous Virchow believed that retinoblastoma belongs to glial tumors, but most modern authors adhere to the theory of the neuroectodermal origin of the tumor. Retinoblastoma develops in any of the nuclear layers of the retina. The tumor cells themselves have a hyperchromatic nucleus and a rather small cytoplasm in volume. There are a large number of mitoses in the cells, and the phenomena of necrosis and apoptosis are pronounced. Calcifications form in the area of cell destruction, which is especially typical for large tumors. Retinoblastoma cells can have varying degrees of differentiation.
Symptoms and complications
More than 80% of retinoblastomas are first discovered by a family member or friend (and only less than 20% by a doctor). Often, due to the child's age, it is difficult for a parent to find out whether the child has retinoblastoma because the child may not report any eye problems. However, it is important for a parent or caregiver to notice any unusual signs that resemble retinoblastoma. Detecting retinoblastoma earlier leads to better outcomes.
Typical signs (more than 20% of cases) include:
- leukocoria - when light enters the eyes, a white reflection appears, giving the impression that the pupil is white;
- strabismus - when one eye does not seem to be looking at the intended target.
Because this condition affects the retina, the child's vision is compromised. Partial or complete blindness in one or both eyes is a possible complication. If detected early, there is a very high survival rate, estimated at 90-95%. Although most children with retinoblastoma have one eye affected, the cancer can spread to the other eye or other parts of the body, such as the brain, lungs, and bones. Metastasis or spread of cancer is rare. If metastasis is present, tumor spread is usually observed 1-2 years after treatment of the original cancer.
Symptoms of retinoblastoma
Depending on the type of tumor growth (exophytic or endophytic), the clinical picture of the disease will also differ. In the case of endophytic growth, retinoblastoma is formed from cells that are located on the inner surface of the retina. With exophytic growth, the source of the tumor is the outer layer of the retina.
Most often, it is possible to detect a neoplasm before the tumor process has left the orbital area. Symptoms of retinoblastoma largely depend on the size of the tumor and its location.
Among the initial symptoms of the disease, leukocoria, that is, the white pupillary reflex (cat's eye symptom), is most often present. Parents perceive this sign as a pathological glow on one or both sides. For this to become noticeable, the retinal tumor must reach a fairly large size. If a tumor dissection of the retina occurs, then a tumor protrudes behind the lens lens, and the tumor itself becomes noticeable. Before leukocoria occurs, even small tumors located near the central retina can cause loss of central vision. With a unilateral process, the patient loses binocular vision, which causes strabismus. With retinoblastoma, vision loss is an early symptom, but it can be difficult to detect because young children may not complain. Strabismus, which is the second most common sign of retinoblastoma, is detected much more often because this symptom is noticeable to others.
The development of retinal cancer is not accompanied by the appearance of pain until secondary glaucoma or an inflammatory process occurs. Pain with retinoblastoma ranks third among all symptoms of the disease.
With late diagnosis of cancer, the process spreads beyond the orbit and often leads to the appearance of metastases. Bilateral tumors of hereditary nature most often have multiple growth foci on both sides. In the case of sporadic mutations, the tumor occurs in only one place.
What is retinoblastoma?
Retinoblastoma is a malignant tumor that develops in the intraocular space from the retinal tissue. This disease occurs in the vast majority of cases in children under 5 years of age, regardless of the gender of the child. This happens for the reason that the neoplasm originates from embryonic tissues. We can say that the disease begins during the period of intrauterine formation of the child. Such children are born in more than 50% of cases from parents who suffered the disease in childhood, i.e. this pathology is of a genetic nature.
Retinoblastoma is located on the retina in the intraocular space
Does retinoblastoma occur in adults? Rarely. Specialists with many years of clinical practice note that over the entire period of work they encounter isolated cases of histologically proven disease in adult patients. It can be assumed that the cause of the development of retinoblastoma in this case is a tumor node that was formed in childhood, but due to unclear circumstances did not develop.
Course of retinoblastoma
If the retinal tumor is located intraocularly, there is a threat to vision. With ophthalmoscopy, cancer can be determined if the node enlarges to 0.25 mm. Externally, such a node looks like a translucent hemispherical mass, which is located within the fundus of the eye and covers the choroid.
As the tumor node grows, it becomes impenetrable and acquires a pinkish color. Sometimes the growth of retinoblastoma is directed into the vitreous body (endophytic tumor). This type of retinal cancer is usually friable and spreads to the vitreous substance and even to the structures of the anterior chamber of the eye. During intraocular tumor growth, secondary glaucoma occurs due to a large tumor size or retinal detachment. At the same time, increased vascular growth occurs within the iris, which leads to an increase in intraocular pressure. In a child, intraocular hypertension can cause an increase in the size of the eyeball, which gradually loses visual function. Later pain, anorexia, and vomiting develop. When the tumor grows beyond the retina, an exophytic variant of retinoblastoma occurs.
When the process spreads beyond the orbit, there is a threat not only to the patient’s vision, but also to his life. This is due to the immediate proximity of the central nervous system. Most often, the tumor penetrates into brain structures through the pia mater or along the fibers of the optic nerve. when the choroid is damaged, we are talking about hematogenous metastasis of retinoblastoma. Sometimes the tumor grows into the sclera. Lymph nodes are also affected, and in later stages distant metastases occur.
Reasons for development
The exact causes of retinal cancer in children have not yet been proven, but in the majority of cases the disease is hereditary.
Retinoblastoma in children can occur due to the following factors:
- harmful environmental conditions, which are observed in large industrial cities,
- poor quality food containing many carcinogens,
- professional activities of parents related to chemicals or other harmful substances,
- age of the parents – the risk of retinoblastoma increases in children if the parents are over 40 years old at the time of the child’s birth.
Most often, retinoblastoma is considered hereditary, but it has a clear connection with the environmental impact on a woman during pregnancy, which affects the fetus. As retinoblastoma grows, it grows beyond the retina and affects the eye socket and optic nerve. Treatment of retinoblastoma in children must be carried out without delay, otherwise the tumor will gradually involve the orbital tissue, and distant metastases will begin to affect vital organs. Parents who have already encountered this pathology are obliged to carefully monitor the development of their children's eyes and regularly visit an ophthalmologist with them.
Differential diagnosis
The doctor must distinguish retinoblastoma from other diseases that cause retinal detachment. Typically, these symptoms are characteristic of vascular anomalies, in which telangiectasia forms in the area of retinal detachment. Also, ultrasound cannot detect signs of a tumor process. However, some pathologies (retinal dysplasia, cicatricial retinopathy) can give an ultrasound picture similar to retinoblastoma.
It is also necessary to distinguish the tumor from granulomas in the vitreous region and retinal hamartomas. They may be associated with inflammation, including damage caused by nematodes. Hamartomas can be combined with tuberculous sclerosis and neurofibromatosis. In parasitic eye lesions (Toxicara canis), the diagnostic features may be very similar to retinoblastoma. In addition, retinoblastoma growing extraocularly must be distinguished from soft tissue sarcomas (rhabdomyosarcomas) and metastases of neuroblastoma. Damage to the eye and orbit can occur against the background of leukemia and lymphomas.
Local (local) chemotherapy
This chemotherapy is indicated for patients from groups C and D in combination with systemic chemotherapy. The technique involves using a superthin catheter to inject a cytostatic drug called Melphalan through the internal carotid and femoral arteries directly into the mouth of the ophthalmic artery. Before this, carotid angiography of the internal carotid artery is performed to visualize the structure of the vessels.
Chemotherapy with Melphalan (selective intra-arterial) is carried out 3-4 weeks after the 1st course of systemic chemotherapy. The dosage is 5-7.5 mg/m2:
- a dosage of 5 mg/m2 is indicated when both eyes are treated simultaneously;
- if only one is affected, then 7.5 mg/m2 should be administered.
16 mcg of Melphalan (this is 0.05 ml) is injected into the vitreous body (med. – “intravitreal”). This concentration is effective against tumor screenings and is safe.
Stages of retinoblastoma
Treatment for retinoblastoma is determined by the size of the tumor and the extent of the pathological process. In this regard, it is very important to correctly stage the tumor.
Among the classifications of retinoblastoma, the one most often used is Reese-Ellsworth, created back in 1969. However, it only includes intraocular tumors. This classification is based on the location of retinoblastoma, the extent of organ damage and the impact of this on the risk of vision loss. All patients with retinal cancer are divided into five groups, which differ in the likelihood of maintaining visual function.
How does non-hereditary (sporadic) retinoblastoma occur?
A non-hereditary form of retinoblastoma appears when two alleles of the retinoblastoma gene are broken in one single cell of the retina, that is, two mutations [mutation] occur in the retinoblastoma gene (in the language of specialists this is called a somatic mutation). In children with this form of the disease, only one tumor almost always grows (doctors call it unifocal retinoblastoma) and only in one eye (specialists call it unilateral retinoblastoma, or unilateral tumor). This retinoblastoma is not inherited.
Reese-Ellsworth classification
Group 1 is the most favorable:
A. A solitary tumor is located at or behind the equator. Has a diameter less than four times the size of the optic disc. B. Multiple nodes, the diameter of which does not exceed 4 disc diameters. Located in the same area.
Group 2 also considers it favorable:
A. A single tumor measuring 4-10 disc diameters, located at or behind the equator. B. Multiple tumors of the same size and location.
Group 3 has a questionable prognosis:
A. A tumor of any size located in front of the equator of the eye. B. A single tumor measuring more than 10 discs behind the equator.
Group 4 has an unfavorable prognosis for vision:
A. Multiple nodes larger than 10 disc diameters. B. Any swelling in the area anterior to the dentate line.
Group 5 has the most unfavorable prognosis:
A. The tumor affects more than half of the retina. B. Contamination of the vitreous body has occurred.
To determine the size of the tumor node, the diameter of the optic nerve (1.5 mm) is used.
There is no uniform staging classification for common retinoblastomas. According to the classification proposed by scientists from the USA, there are:
- The first stage is characterized by the location of retinoblastoma only within the retina.
- The second stage is accompanied by the spread of the tumor to other structures of the eyeball.
- The third stage has extraocular spread of the tumor.
- At the fourth stage, distant metastases of retinoblastoma are present.
Because this classification does not include important characteristics of retinal cancer, some doctors prefer to use a different classification to guide their treatment of the disease. There are several variants of tumor growth that have prognostic significance:
- Intraocular location of retinoblastoma.
- Tumor damage to the optic nerve fibers.
- Involvement of orbital tissue in the process.
- Formation of distant metastases.
Causes
The body's cells reproduce throughout life—tissues wear out and cells are replaced in a controlled manner. Retinoblastoma, like all cancers, occurs when this control is lost and cells begin to divide at an unusually high rate. Researchers are still working on the exact causes of the gene mutation in retinoblastoma. However, they found an inherited link.
Retinoblastomas can be divided into 2 categories:
- one is caused by inheritance of a defective gene,
- and the other is a random change in the RB gene during cell replication (sporadic form).
It is the sporadic form that is the most common. About 90% of children with retinoblastoma have no previous family history of the disease. Meanwhile, children who have a parent with the disease make up less than 10% of all diagnosed cases.
Treatment of retinoblastoma
To treat retinoblastoma in modern oncology, surgical removal, chemotherapy, and radiation therapy are used. To obtain satisfactory results, a combined approach is recommended. When choosing a tactic, the following parameters of retinoblastoma should be taken into account:
- Is vision preserved?
- Pair of lesions;
- Extension of the process beyond the eye;
- Involvement of the orbit and central nervous system in the tumor process, formation of distant metastases.
Doctors try in every possible way to limit themselves to conservative methods. Among them, photocoagulation and cryotherapy have become very popular. These techniques rarely cause complications and help preserve vision. If the disease recurs, this minimally invasive procedure can be repeated. Cryotherapy is used when retinoblastoma is located in the anterior retina; photocoagulation is more suitable for tumors of the posterior retina.
What happens after treatment?
Depending on the treatment chosen, the child may experience side effects. For example, children treated with chemotherapy may experience nausea, weakness, dizziness, or fever. And in the case of radiation treatment, redness or dryness of the irradiation area may occur.
After treatment is completed, the child can return to normal life provided he feels well and has the doctor's permission. Recovery time varies depending on the treatment method and the stage of the disease. In cases where it is impossible to save a child’s eye, clinics create high-quality prostheses that do not differ in appearance from a real eye.
Surgical method
For retinoblastoma, surgery usually involves removing the eye (enucleation). This operation is indicated for massive intraocular lesions, the presence of serious glaucoma, absence of vision and the impossibility of restoring it. During the operation, it is necessary to cut off the optic nerve as far from the eye as possible.
Enucleation gives good results for the patient's life (mortality is minimal). Also, the patient no longer requires frequent examinations under anesthesia.
Due to the fact that enucleation is performed more often on young children (less than three years old), serious cosmetic defects may occur. This is due to the absence of a formed orbit and the subsequent active growth of certain structures.
After removal, the eyes are sent for histological examination. When tumor cells invade the fibers of the optic nerve, in case of tumor growth into the choroid, the prognosis is unfavorable. When retinoblastoma grows extraocularly, orbital exenteration is performed, which is even more traumatic than enucleation.
Enucleation for retinoblastoma
The most effective treatment for eye cancer is enucleation - removal of the eyeball. During the operation, the optic nerve is cut as far as possible, so this method shows the best results in the treatment of retinoblastoma. 6 weeks after surgery, prosthetics can be performed to eliminate the visual defect. Because patients with retinoblastoma are often children, enucleation poses some challenges. After removal of the eyeball, the bones of the orbit begin to grow more actively, causing a severe defect.
Indications for enucleation:
- extensive damage and inability to restore vision,
- massive seeding of the vitreous body,
- the tumor affects the posterior chamber of the eye and reaches the posterior capsule of the lens,
- localized near the ciliary body or occupies most of the anterior part of the eye,
- glaucoma due to vascular proliferation,
- advanced secondary complications (uveitis, cataracts, hemophthalmos, hyphema),
- the onset of blindness with the inability to restore vision.
After enucleation, the eyes are sent for histological analysis. If the tumor has affected the optic nerve and choroid or has extended beyond the sclera, this indicates a poor prognosis for the patient. When retinoblastoma extends beyond the orbit, orbital exenteration (removal of orbital contents) is recommended.
In what cases are enucleation and conservative treatment combined:
- retinoblastoma grows into the optic nerve,
- scleral damage,
- extrabulbar tumor spread,
- location in the peripapillary zone,
- the presence of large and multiple tumor nodes,
- penetration of lesions into the vitreous body, iris, anterior segment of the eye,
- choroidal involvement.
Radiation treatment
To try to preserve vision in patients with retinoblastoma, radiation is given to the tumor. The goal of this technique is to cure the disease and preserve vision. In this case, the doctor must assess all the risks of immediate and long-term results.
The technique of radiation therapy can be different, but external irradiation (two lateral fields) is most often used. Due to the frequent occurrence of bilateral retinal involvement and multifocal tumor growth, the training field should cover all these areas. In addition to the retina, radiation therapy irradiates the vitreous body and 10 mm of the anterior segment of the optic nerve. Doses depend on the stage of the disease (at stages 1-3 - 3500 Gy, at stages 4-5 - 4500 Gy). Special blocks are used to protect the lens. This helps prevent the formation of post-radiation cataracts. If, after lateral irradiation, tumor recurrences occur in the anterior segment, cryotherapy is additionally prescribed.
During irradiation, young children are put under anesthesia or their head is firmly fixed. With this treatment, about 75% of patients are cured, which is due to the high sensitivity of the tumor to radiation therapy. If cryotherapy is additionally performed, the percentage of cured patients increases even more.
Sometimes radioactive plates are used, but this technique is limited. This is due to the uneven distribution of the radiation dose. Vascular damage and hemorrhage may occur at the site of greatest activity.
Radiation therapy is most often used for tumor processes affecting the central zones of the retina, as well as for solid retinoblastomas.
Treatment methods
With timely detection of retinoblastoma of the eye, with adequate treatment, in 90% of cases the prognosis for children is favorable. Treatment methods for retinoblastoma vary, and when choosing the most optimal method, ophthalmologists take into account the following factors:
- preservation of vision, as well as the chances of its recovery after treatment,
- number of affected eyes,
- retinoblastoma size,
- the tumor is located on or behind the equator,
- involvement of the optic nerve, spread of the process to the tissues of the orbit,
- the presence or absence of distant metastasis.
During treatment, not only the ability to preserve vision, but also the safety of the eye itself is of particular importance. After surgery to remove an intraocular tumor, there may be a significant disruption in the growth of the skull in the facial area, which can cause serious cosmetic defects. Maximum preservation of eye tissue is possible with drug treatment of retinoblastoma, but it is rational only at the beginning of the development of the disease. For bilateral lesions, a separate treatment method is selected for each eye.
Cryotherapy and photocoagulation
These methods can be rationally used in the first stages of development, while they allow preserving not only the organ, but also visual function. After several sessions of cryotherapy, small tumors located on the anterior periphery can be eliminated. Photocoagulation allows you to remove small tumors that form on the retina. After such treatment, there is still a risk of retinoblastoma recurrence, but in such a situation, you can undergo another course of treatment.
Surgery
During surgical procedures, not only the eye, but also the orbital tissue can be removed. This operation is prescribed if the following indications exist:
- extensive damage to the organ, when the use of conservative methods does not produce results,
- development of glaucoma due to tumor growth,
- large tumor size,
- loss of vision without the possibility of its restoration.
During surgical procedures, it is very important that the optic nerve is cut off as far as possible from the affected area. This is necessary to prevent the spread of neoplasia. Some time after the operation, it is possible to install a prosthesis.
Radiation therapy
Radiation therapy can be carried out remotely and using applications of radioactive substances. It is rational to use irradiation for small tumors in children located outside the optic nerve. If a child suffers from congenital retinoblastoma, he is prescribed external irradiation, which allows one to influence several foci of pathology at once.
In approximately 70% of cases, irradiation can achieve a positive result and even preserve vision. Radiation therapy is also combined with surgery and drug treatment. The radiation dose is usually about 4500 Gy. In addition to the retina, the vitreous body and 10 mm of the anterior part of the optic nerve are irradiated.
Chemotherapy
Indications for a course of chemotherapy are:
- extensive damage to the tissues of the orbit,
- optic nerve damage
- tumor metastasis.
During treatment, patients are prescribed special medications that can be administered intravenously or orally. This method is quite effective for treating children with diagnosed retinoblastoma, but chemotherapy has a large number of side effects, including hair loss, nausea, apathy and general malaise.
Folk remedies
It is impossible to get rid of retinoblastoma using traditional methods. Folk remedies include the use of decoctions and teas that help relieve tension and weaken the clinical manifestations of the disease. Also, after a course of chemotherapy, acupuncture is recommended, which can improve the general condition of the patient. Before using any alternative medicine, you should always consult a doctor.
Chemotherapy
After surgical treatment and radiation therapy, almost 80-90% of patients are cured. Chemotherapy is justified in cases of massive lesions, invasion of tumor cells into the fibers of the optic nerve (in particular in the resection zone), in the case of regional metastases and orbital damage.
For chemotherapy, a number of cytostatics are used in combination (vincristine, carboplatin, Vepezid). Other combinations of drugs are also used (vincristine, dixorubicin, cyclophosphamide). However, cyclophosphamide increases the risk of secondary tumors and causes sterilization of patients.
Due to the development of diagnostic equipment in developed countries, the number of patients with early stage retinoblastoma is increasing. Therefore, photocoagulation, cryotherapy and radiation therapy are usually sufficient for cure.
Prognosis and prevention
It is very good when retinoblastoma can be detected in children at the very beginning of their growth. Then, in addition to surgery, it is possible to carry out other treatment methods to preserve the organ of vision: cryotherapy, photocoagulation and radiation therapy. When metastases appear, retinoblastoma is removed by enucleation of the eye, which allows the child to be completely cured. However, the loss of the eye leaves a significant cosmetic defect. If, as retinoblastoma grows, secondary lesions appear in the brain, the chances of saving the child’s life are significantly reduced.
The main reason for the development of a disease such as retinoblastoma in children is genetic mutations that are transmitted hereditarily. Therefore, if one of the parents has already had retinal cancer, the family is required to undergo medical genetic counseling. Children at risk should undergo a full medical examination from an early age.
Malignant tumors (retinoblastoma) | Intraocular tumors
Description
The most common malignant intraocular tumors in children are retinoblastomas.
They account for 89.3% to 98.2% of intraocular tumors in childhood. Melanomas of the vascular tract in childhood, especially in preschool, primary and secondary school, are extremely rare. Retinoblastoma
- intraocular malignant tumor of the retina, occurring mainly in early childhood. The tumor is quite rare - it occurs in approximately 1 in 15,000-34,000 newborns.
In the structure of eye morbidity, tumors range from 0.001 to 0.008%, in the structure of childhood eye morbidity - 0.07%.
The proportion of patients with retinoblastoma among patients in large children's ophthalmology hospitals ranges from 0.06% to 4.5%, and among hospitalized children with tumors - up to 41.3%. Retinoblastoma
is a congenital tumor that can be found in newborns. Retinoblastoma has even been observed in fetuses. In this regard, many authors suggest that the tumor arises in utero and is not clinically manifested for a long time and, therefore, is not diagnosed.
Clinically, retinoblastoma manifests itself at various times after birth. As a rule, it occurs in children under 6 years of age (97%), most often under the age of 3 years (70%). In 22-23% of patients, retinoblastoma is detected in the first year of life. In children over 6 years of age, gore is rarely observed (in adults - as a casuistry).
Boys and girls get sick equally often. Bilateral retinoblastoma is observed in approximately 20-40% of patients and occurs mainly in children under 3 years of age. Damage to the second eye may not occur simultaneously with the disease of the first, sometimes after several years.
Unilateral retinoblastomas usually develop as a result of somatic mutations affecting only the retinal cells. Bilateral retinoblastomas are often the result of germ cell chromosome mutations. Hereditary forms of the tumor are observed in approximately 40-50% of patients.
Healthy parents who have one child with retinoblastoma have about a 6% chance of developing the tumor in subsequent children. In cases where several children in a family are sick, the risk of developing retinoblastoma in subsequent children is 50%. More than half of children whose parents had retinoblastoma develop this tumor.
According to histological structure, retinoblastoma is a malignant peiroectodermal tumor that develops from nerve cells of the embryonic retina. The histogenesis of the tumor has not been definitively established. The tumor can develop from any part of the optical part of the retina.
Retinoblastoma consists of undifferentiated neuroblastic cells, retinoblasts, which are characterized by large hyperchromatic nuclei, scanty cytoplasm, and a large number of mitoses. There is no stroma in the tumor.
Depending on the degree of differentiation of tumor cells, a distinction is made between retinoblastoma, which is more common, and retinocytoma. The more malignant form is the undifferentiated form, retinoblastoma. In this form, the tumor cells are less differentiated and group around the vessels, forming so-called pseudorosettes.
Differentiated form of tumor
- retinocytoma - has a more favorable course. Retinocytoma consists of more differentiated cells from which true Flexner-Wintersteiner rosettes are formed. The tumor may have a mixed structure.
There are two types of retinoblastoma growth: exophytic, or creeping, and endophytic with tumor prominence into the vitreous body. Newly formed vessels develop in a growing tumor. Tumor cells proliferate around the neoplasm's vessels.
Due to insufficient blood supply, the tumor undergoes necrosis quite early, the foci of which are the source of dispersion of individual cells and entire conglomerates during endophytic growth into the vitreous body, and during exophytic growth into the subretinal space and choroid. Calcium salts are deposited in foci of necrosis, forming calcifications characteristic of a tumor.
The clinical picture of retinoblastoma is varied and depends on the stage of development of the process.
Stage I
is characterized by the intraocular location of the tumor. Initially, the tumor is located within the retina; as it grows, it disrupts the vitreous plate and spreads to the choroid and vitreous body.
Initially, a grayish, slightly cloudy flat lesion appears in the fundus. As the tumor grows, a gray-whitish-yellowish node of a round or oval shape with clear boundaries, a smooth or bumpy surface, which can be covered with the retina with vessels passing through it, is formed that protrudes into the vitreous body. Retinoblastoma nodes can have different localizations and be single or multiple. The latter can merge with each other in the process of growth. The rapid growth of a tumor with disruption of its metabolic processes and blood supply leads to early necrosis; foci of necrosis with cheesy decay undergo calcification.
Due to the peripapillary growth of the tumor, compression of the retinal vessels occurs, congestion develops, accumulations of subretinal transudate are formed, and retinal detachment occurs, initially limited, and then total (Fig. 135).
Visual acuity when the tumor is located in the central parts of the fundus of the eye decreases early and sharply, and strabismus appears. In cases where the neoplasm is localized in the periphery, vision decreases as it spreads to the central region.
Stage II
characterized by further intraocular tumor growth, intraocular dissemination of tumor cells, and secondary glaucoma.
At this stage, dissemination of tumor cells and their conglomerates into the vitreous body and anterior chamber is observed, which occurs during the disintegration of tumor tissue, where a pseudohypopyon is often formed and precipitate-like deposits appear on the posterior surface of the cornea. As a result of dissemination of tumor cells, grayish tumor nodules can form on the surface of the iris.
The tumor, increasing in size, fills the entire cavity of the eyeball and grows into the choroid. As a result of destruction and growth of the trabecular apparatus of the eye, the outflow of intraocular fluid is disrupted and intraocular pressure increases.
Pain in the eye, stagnant injection, swelling of the cornea appears, pupil dilation and lack of reaction to light are observed. In young children, under the influence of increased ophthalmotonus, the eyeball usually enlarges (buphthalmos). With extensive dystrophic changes and necrosis of tumor tissue, a toxic inflammatory process (iridocyclitis, uveitis) often develops.
In these cases, newly formed vessels in the iris and posterior adhesions appear. Exophthalmos may occur due to swelling of the orbital tissue.
Stage III
retinoblastoma is characterized by extraocular spread of the tumor, which grows beyond the eyeball along the optic nerve, scleral emissaries, and through the sclera. Along the optic nerve, the tumor spreads along its fibers and under the arachnoid membrane; after germination of its dura mater, retinoblastoma infiltrates the tissues of the orbit.
The spread of the neoplasm into the orbit is accompanied by rapidly increasing exophthalmos and, due to infiltrative growth, can destroy the bone walls of the orbit and grow into the paranasal sinuses. When the tumor spreads along the optic nerve into the cranial cavity (into the subarachnoid space), brain symptoms (headache, nausea, vomiting) are observed.
The growth of retinoblastoma through the wall of the eyeball leads to the formation of an extrabulbar neoplasm. When the tumor spreads anteriorly, foci of corneal necrosis appear, and when it grows through the cornea or sclera, an extrabulbar node is formed, which sometimes reaches significant sizes.
IV stage
retinoblastoma (metastasis stage) is characterized by the presence of lymphogenous and hematogenous metastases. Lymphogenously, retinoblastoma metastasizes to the lymphatics, mainly to the parotid, submandibular and cervical nodes, and hematogenously to the skull bones, tubular bones of the lower extremities, and liver.
In older children, retinoblastoma often has a somewhat atypical course. The clinical picture may resemble manifestations of uveitis. In these cases, tuberculous etiology of the process is often suspected. In children of this age, an infiltrative form of the tumor is often observed, characterized by diffuse thickening of the retina, accumulation of exudate in the anterior parts of the vitreous, early appearance of pseudohypopyon and anterior adhesions.
In rare cases, spontaneous regression of retinoblastoma may occur, the development of which is associated with immunological factors and circulatory disorders in the tumor.
The classification of retinoblastomas by TCFM (1982) allows for a more detailed grouping of tumors into stages depending on the degree of damage, which is important for the clinical characteristics of the tumor, assessment of prognosis and analysis of treatment results.
The degree of damage to the organ of vision by the tumor is assessed on the basis of clinical data (preoperative clinical classification) and the results of histological examination of the tumor after removal of the eye (postsurgical histomorphological classification). The TCM classification makes it possible to assess the condition of lymph nodes and distant metastases.
Each eye should be assessed separately. The classification does not reflect complete regression of the tumor. After enucleation, it is necessary to carry out histological verification. Each unusual case must be considered separately.
Additional research methods - radiography, echography, computed tomography, scanning, bone marrow puncture, cerebrospinal fluid examination - help clarify the diagnosis.
CLINICAL EVALUATION CATEGORIES
Diagnosis of retinoblastoma is often very difficult. The diagnosis is established on the basis of medical history, the results of ophthalmological, laboratory and instrumental studies and general clinical examination.
The first signs of retinoblastoma, which ophthalmologists, pediatricians, paramedics, and adults around the child pay attention to, are the following:
1) leukocoria
- a whitish-yellowish glow of the pupil due to the reflection of light from the surface of the tumor (see Fig. 135).
With the central location of small tumors, the glow of the pupil is detected when looking straight; with retinoblastomas located on the periphery of the fundus, the glow of the pupil is visible at the extreme abductions of the eye. In the presence of large tumors that occupy a large area of the fundus or occupy the entire or almost the entire posterior part of the eyeball, the glow of the pupil is visible in all directions of view;
2)
dilation of the pupil, weakening of its direct reaction to light;
3)
strabismus (the last two symptoms are caused by decreased vision due to the presence of a tumor). In cases where these signs are present, it is necessary to conduct a thorough examination of the child’s fundus, having previously achieved maximum pupil dilation using mydriatic agents, and, if indicated, perform a detailed examination.
Diagnosis of retinoblastoma is based on the clinical picture of the disease, however, the results of an ophthalmological examination must be compared with data obtained from laboratory and instrumental examination and a general clinical examination of the child.
Ophthalmological examination in children with retinoblastoma includes examination with side illumination, ophthalmoscopy, biomicroscopy, tonometry, etc. Examination of young children using the necessary research methods is carried out under narcotic sleep conditions.
One of the most informative methods is ultrasound dowsing, which allows, by identifying plus tissue, to distinguish retinoblastoma from endophthalmitis, retinal detachment, retrolental fibroplasia, and vitreous fibrosis. At the same time, in patients with Coats' retinitis, using conventional ultrasound examination techniques, signs similar to those of retinoblastoma can be detected.
The use of quantitative echography in these cases, as well as determination of the amount of ultrasound attenuation in the tumor, expands diagnostic capabilities. Retinoblastoma is characterized by ultrasound attenuation ranging from 0.2 to 1.0 dB per 1 mm of tumor tissue. An attenuation value below 0.2 dB per 1 mm indicates the non-tumor nature of the plus tissue. Ultrasound echobiometry is important for obtaining data on the dynamics of the process, allowing one to determine the degree of tumor prominence.
X-ray diagnosis of retinoblastomas is based on identifying foci of calcification in the tumor tissue, which appear as fine-grained shadows on an x-ray. In stages III and IV of the disease, radiographs can reveal darkening and widening of the orbit, thinning and defects of its walls, widening of the optic nerve canal, and darkening of the paranasal sinuses.
In unclear cases, a radioisotope study is indicated, based on the accumulation of radioactive isotopes in the tissue of retinoblastoma; it often allows a correct diagnosis to be made. Due to the absence of pigment in the retinoblastoma tissue, diaphanoscopy is usually not very informative.
Immunological and biochemical methods are promising. In children with retitioblastoma, a decrease in a number of indicators of humoral and cellular immunity is observed. The content of total glycoproteins in the blood serum and the excretion of glycosaminoglycans in the urine in retinoblastoma exceeds normal values. (General clinical examination of children with retinoblastoma allows us to exclude the presence of chronic infections (tuberculosis, toxoplasmosis, syphilis) and other diseases.
In order to detect possible metastasis, other organs are examined. Computed tomography allows in some cases to detect the growth of retinoblastoma into the brain. X-rays are used to determine bone metastases.
Scanning helps detect liver metastases. To clarify the diagnosis of distant metastases, if indicated, a bone marrow puncture is performed, as well as a spinal puncture, followed by a cytological examination of the cerebrospinal fluid, in which, in cases of retinoblastoma germination into the subarachnoid space, tumor cells can be detected.
Differential diagnosis of retinoblastoma often presents significant difficulties. The tumor should be differentiated from Coats' retinitis, endophthalmitis, retrolental fibroplasia, vitreous fibrosis, retinal detachment, Hippel-Lindau disease, metastatic ophthalmia, chronic inflammatory processes, parasitic diseases, etc. Among parasitic diseases, special attention is paid to toxoplasmosis and larval granulomatosis caused by Toxocara canis .
Treatment of retinoblastoma, given its rapid growth and tendency to metastasize, must begin immediately after diagnosis.
For retinoblastomas, combined treatment is used, including surgery, radiation therapy, chemotherapy, photo and laser coagulation, etc. The treatment method and the set of methods used depend on the stage of the tumor, as well as on the symmetry of the lesion (unilateral or bilateral).
For unilateral lesions, enucleation is performed; for bilateral lesions, the more affected eye is removed, and the other is treated conservatively. Due to the danger of tumor growth into the optic nerve, its intersection during enucleation is carried out at a distance of at least 10-15 mm from the posterior pole of the eyeball.
With extrabulbar spread of retinoblastoma, orbital exenteration is indicated.
After surgical intervention, the question of further tactics and treatment methods are decided on the basis of the results of a histological examination of the removed eye. In the presence of a small tumor (T1 - T2 N0M0) stage I and 1a, located outside the peripapillary region, enucleation can be limited to enucleation in patients with unilateral retinoblastoma.
Conservative treatment after eye removal is indicated for:
- tumors of stages IIIa and IIIb - germination of retinoblastoma into the optic nerve, along the emissaries of the sclera and extrabulbar spread of the tumor in other ways;
- retinoblastomas of stages I, IIa, IIb and IIc in the presence of large or multiple (t) tumor nodes, intraocular dissemination of the process (into the vitreous body, iris, anterior chamber, etc.), location of the tumor in the peripapillary region, involvement of the optic nerve head in the process and choroids.
For stage IV retinoblastoma (all T, N, M), palliative treatment (chemotherapy, symptomatic therapy) is carried out in a specialized institution.
The most radical operation for retinoblastoma is enucleation, however, due to the success of conservative treatment, it is considered possible to use organ-preserving treatment not only for unilateral, but in some cases also for bilateral (preserving both eyes) tumors.
Conservative treatment, including radiation therapy, chemotherapy, photo-, laser- or cryocoagulation, is carried out according to strict indications: for tumors of stages I and Ia (small single tumor nodes without intraocular dissemination, located outside the macular and peripapillary region). The dynamics of tumor changes under the influence of treatment are judged by the results of ophthalmoscopic and ultrasound examinations.
X-ray therapy, radioactive isotopes (?-application), etc. are used as radiation therapy. Remote radiotherapy irradiates the orbit of the removed eye, and in case of bilateral tumors, the preserved eye is also irradiated. Irradiation is carried out daily, starting from the 2nd - 3rd day after surgery; a single exposure dose is 200 R, the total dose for a series is 3500-4000 R.
The course of treatment includes one or two series of radiation, which are carried out with an interval of 1/2-2 months. To prevent radiation keratitis and cataracts of the irradiated eye with bilateral retinoblastoma, instill a 2% freshly prepared cysteine solution (or 1% taufon solution) before each irradiation session (2 drops 6 times every 15 minutes). v-Therapy is indicated in cases of massive tumor growth into the orbital cavity after exenteration.
The indication for the use of ?-applicators is stage I retinoblastoma in the presence of a single tumor node of a relatively small size with a prominence of up to 5–7 mm. The advantage of γ-therapy is that it allows a large dose of radiation to be delivered to the tumor without damaging the surrounding healthy tissue.
Metal ophthalmic strontium P-applicators (strontium + yttrium) are used. The R-Applicator is sutured to the sclera in the area corresponding to the location of the tumor; it is removed after 2-4 days. Applicators with radioactive cobalt and ruthenium are also used.
Electronic therapy is promising, which makes it possible to create a high concentration of energy at a predetermined depth at low input and output doses.
Chemotherapy is combined with surgery and radiation therapy. Cytostatic drugs are used as chemotherapeutic agents: prospidin, fopurine, cyclophosphamide, as well as thiophosphamide, embiquin, etc. Chemotherapy drugs are administered intramuscularly, intraarterially, subconjunctivally and retrobulbarly.
Photocoagulation, as a rule, is carried out in combination with other methods (radiation therapy and chemotherapy, etc.) at the final stages of treatment. It is recommended for use in small tumors of stage I retinoblastoma (up to 25% of the fundus area and node protrusion no more than 8 mm).
In order to disrupt the blood supply to the tumor and create a chorioretinal barrier, a double shaft of coagulants is first created around the tumor node. After 3-4 weeks, destructive coagulation of the tumor tissue begins. Two to seven sessions are performed at intervals of 3-4 weeks, during which swelling and hemorrhages resolve. Photocoagulation is contraindicated for tumors located near the optic disc and macular region, as well as for large tumors.
Argon laser light coagulation can be used to treat stage I retinoblastomas in the presence of small tumor nodes. For bilateral retinoblastomas, for the treatment of the remaining eye, laser coagulation is used in combination with other methods, most often with radiation therapy and chemotherapy, usually in the final stages of treatment.
The advantage of the method is the absence of a pronounced perifocal reaction of the tissues surrounding the tumor, which makes it possible to perform laser photocoagulation for tumors located near the optic nerve.
Transscleral diathermocoagulation is aimed at destroying the vessels feeding the tumor and coagulating the tumor tissue. The method is used in combination with photocoagulation. Due to its low effectiveness and the development of a pronounced postoperative reaction, indications for this method are limited mainly to cases where other methods have proven ineffective.
Cryocoagulation is used in combination with photocoagulation. Liquid nitrogen and carbon dioxide are used as a cooling agent. The advantages of the method over diathermocoagulation are less damaging effects on the sclera. Cryosurgery is considered indicated for tumors ranging in size from 0.5 to 7.0 times the diameter of the optic disc, protruding by 3.0-3.5 mm.
According to modern authors, the mortality rate for retinoblastoma is 15%. However, the prognosis and results of treatment for retinoblastoma largely depend on the stage of the tumor, as well as on the symmetry of the process (unilateral or bilateral).
The prognosis is more favorable for stage I-II retinoblastomas resulting from somatic mutations. For unilateral retinoblastoma of stage I-II, surgical or combined (according to indications) treatment can save the lives of 98% of patients; for stage III tumors after combined treatment, patient survival is 67.5%. Half (51.1%) of children with stage I-III bilateral retinoblastoma (according to the condition of the more affected eye) manage to save life, and often vision, with the help of combined treatment.
The prognosis for preservation of life and vision is serious, especially with bilateral tumors.
In the long term after radiation therapy, various tumors (usually sarcomas) can develop both in the irradiation zone and outside it.
Dispensary observation of children with retinoblastoma after completion of treatment is carried out in the eye office of the clinic and oncology clinic at the place of residence. The ophthalmologist should conduct an examination once every 2 months during the first year after the end of treatment, once every 3 months during the 2nd year, once every 6 months for the next 2 years, and then once a year.
Patients are not removed from the register.
Children who have had retinoblastoma should be systematically referred for consultation to the medical institution where they were treated. Young children born in families with patients with retinoblastoma should also be under clinical observation.
Diagnosis and prognosis of retinoblastoma
Children with a hereditary predisposition to this disease should be registered with an ophthalmologist.
An ophthalmologist examines the patient when identifying symptoms of the disease: strabismus, leukocoria, enlarged eyeball. For this purpose, biomicroscopy, ophthalmoscopy, gonioscopy, vision examination, measurement of the strabismus angle, and measurement of eye pressure are used. If vision is blurred, an ultrasound of the eye is checked to detect a tumor. Intraocular biopsy can provoke the development of a tumor, as a result of which this procedure is used only in exceptional cases. To determine the extent of the spread and development of the tumor, radiography of the eye space and paranasal sinuses, CT and MRI of the brain, and liver scintigraphy are used.
If the disease is detected at the initial stage, treatment takes place without any losses using radiation therapy and cryotherapy.
Based on the degree of development and its prevalence, procedures are prescribed that can lead to the loss of an eye. The prognosis for detecting this pathology is favorable in most cases. However, treatment methods involving eye exenteration lead to a cosmetic defect and subsequently psychological trauma to the child.
Do you want to know the cost of cancer treatment abroad?
* Having received data about the patient’s disease, the clinic representative will be able to calculate the exact price for treatment.
Symptoms
As retinoblastoma spreads, the following symptoms will appear:
- gradual decrease in vision;
- increased pressure inside the eyes;
- the appearance of pain. Since a child of two or three years old cannot independently express certain sensations, parents can notice the first expressions of signs, paying attention to the fact that their child constantly rubs one eye. Less often, parents can independently detect a decrease in visual acuity;
- a sign of “cat’s eye” - the appearance of a white spot on the pupil;
- loss of binocular vision;
- development of strabismus;
- photophobia;
- increased lacrimation;
- swelling and redness of the eye;
- change in iris color;
- pupil deformation;
- inflammation of the choroid.
Retinoblastoma in a child
The appearance of the following symptoms indicates that retinoblastoma is spreading to distant areas:
- severe intoxication of the body;
- decreased or complete lack of appetite;
- general weakness;
- attacks of nausea and vomiting;
- severe headaches.