Tapetoretinal retinal abiotrophy is a hereditary disease that is accompanied by retinal dystrophy. In this case, the rod photoreceptor apparatus is mainly affected. This pathology does not occur often.
Symptoms of the disease were described back in 1857 by Dr. Doners. He called this disease retinitis pigmentosa. Subsequently, the causes of this pathology were studied in more detail, and the name of the disease was transformed into primary taperetinal dystrophy. This disease is also called rod-cone dystrophy, since in the early stages of the pathology the rod apparatus is affected, and later damage to the cones also occurs. The essence of the disease is reflected in most detail by the term retinal pigment abiotrophy.
The photosensitive layer of the retina of the eye contains two types of photoreceptors: cones and rods. Photoreceptors received such names because of the peculiar shape of the cells. The central zone of the retina contains mainly cones, which are responsible for clear and color vision. Rods are mainly located in the peripheral areas of the retina, but a small number of these photoreceptors are also present in the central zone. Rods are responsible for peripheral vision and also help a person navigate in low light conditions.
As a result of mutation of a number of genes that ensure normal nutrition of the retina, as well as good functioning of this layer, gradual destruction of photoreceptors occurs. The pathological process begins in the peripheral areas, and then gradually moves to the center of the retina. The disease usually takes several decades to progress.
Usually, bilateral eye damage occurs, and symptoms of the disease become noticeable at an early age. By about the age of twenty, vision loss progresses, and patients are often unable to work. In some cases, not the entire surface of the retina is affected, but only a separate sector of it. Sometimes taperetinal abiotrophy of the retina manifests itself at a later age.
Patients with this pathology have a high risk of developing macular edema of the retina, the formation of glaucoma, and cataracts.
Etiology and pathogenesis
The disease is based on a congenital defect in the genetic code. Initially, only the rods are affected, but then the cones are also damaged. Research by geneticists has allowed scientists to identify genes responsible for the occurrence of various forms of retinal pigmentary dystrophy. Symptoms of the disease vary depending on its duration and form. Retinitis pigmentosa leads to blindness and visual impairment.
The worldwide prevalence of retinitis pigmentosa is 1 in 5,000 people. The disease is quite common, there are about 3 million patients and 99 million people who have a gene defective for retinitis pigmentosa.
The age at which the disease occurs, the timing of progression, the prognosis of visual functions and ophthalmological manifestations of retinitis pigmentosa are determined by the type of inheritance.
Inheritance can be sex-linked (passed from mother to son with an X chromosome), autosomal recessive (disease genes are passed on from both parents) or autosomal dominant (pathological gene is passed on from one parent). Since the X chromosome is most often involved, men are affected more often than women.
Patients suffering from retinitis pigmentosa who wish to have children, as well as healthy people with a burdened hereditary history, need to consult a geneticist at a family planning center to determine the prognosis of the visual condition of future offspring. Currently, there are tests to identify carriers of this disease.
Treatment of the disease
Initial treatment for retinal degeneration most often consists of medications . Their main effect is to improve metabolism in the retinal layer, restore the retina and dilate blood vessels. These are emoxipine, mildronate, taufon, etc. These drugs can be administered to the body, both in the form of eye drops and by injection. It is also advisable to use a complex of nucleic acids in treatment - Encad, which in more than half of the cases significantly improves visual functions. It is prescribed intramuscularly, subconjunctivally, using iontophoresis, or local applications are made with it.
Often, in parallel with drug treatment, physiotherapeutic measures , the purpose of which is to stimulate regenerative processes in the retina and activate the remaining rods and cones. Electrical stimulation of the eye, magnetic resonance therapy, and ozone treatment are widely used. Vasoreconstructive surgeries can be used to restore the vascular bed.
Surgical treatment of retinal pigmentary degeneration is used to normalize the blood supply to the retina; for this purpose, some eye muscles are transplanted into the suprachoroidal space.
Recently, encouraging data have been coming from genetic engineers who have found the ability to restore damaged genes responsible for the development of this disease. In addition, special implants have been developed - retinal substitutes.
And very recent experiments on mice conducted in Britain provide convincing evidence that blindness can be treated with the help of special light-sensitive cells injected. And although this technique has not yet been tested in humans, scientists hope that this drug can be used to treat people suffering from retinitis pigmentosa.
As for the prognosis of the disease, it is generally unfavorable, but with early detection of pathology and timely initiation of treatment, the process can be delayed and even an improvement in the condition can be achieved. All patients are advised to avoid prolonged stays in dark rooms and not engage in heavy physical labor.
Symptoms
The most common and early symptoms of the disease are significant deterioration of vision at night and in poorly lit rooms, the so-called “night blindness” (hemeralopia - “night blindness”) and progressive changes in the peripheral visual field.
Violation of dark adaptation is the first symptom and usually occurs several years before changes in the retina determined by an ophthalmologist. Reduced dark adaptation, especially in the early stages of the disease, requires careful investigation.
Causes
It is known that the membrane of the eyes, which is highly photosensitivity, includes two types of cells:
- sticks;
- cones.
The location of the cones is the center of the retina. They are responsible for the level of visual acuity and color perception. The rods are “scattered” throughout the retina, more in its peripheral part than in the center. Thanks to rods, a person has the function of peripheral vision and can perceive objects in poor lighting.
Certain genes are responsible for the nutrition and functions of the retina. If they are damaged, its outer layer begins to gradually collapse. As a rule, the pathological process originates in the periphery, spreading further over time. However, it can last for many years, moving towards the center of the retina.
Abiotrophy is characterized by bilateral eye damage. Primary symptoms are detected in patients already in childhood, and since the manifestations gradually increase, at the age of about 20 years, complete loss of ability to work is likely.
However, in clinical practice several possible variants of the course of the disease have been described:
- It happens that only one eye is affected;
- only a limited field of the retina is affected;
- the illness begins later than usual.
Patients suffering from abiotrophy belong to risk groups with a high probability of developing other ophthalmological pathologies - these are glaucoma, central retinal edema and cataracts.
Free consultation on training issues
Our consultants are always ready to tell you about all the details!
Diagnostics
Diagnosis of retinitis pigmentosa in the early stages of the disease and in childhood is difficult and is usually possible only in children aged 6 years and older. The disease is often discovered when a child has difficulty orienting himself at dusk or at night. Symptoms of retinitis pigmentosa, especially in children, include headaches and flashes of light before the eyes. Diagnosis requires special modern electrophysiological studies (recording of retinal biopotentials). Perimetric studies allow you to assess the condition of the visual fields.
Until the age of 20, most patients with retinitis pigmentosa have visual acuity greater than 0.1, and every fourth patient retains the ability to read, using assistive devices. By the age of 45–50, visual acuity is less than 0.1 and the ability to read is almost completely lost.
Diagnosis of the disease
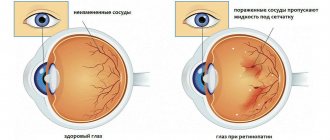
An ophthalmological examination includes testing visual acuity and visual field testing. The structures of the fundus are examined, where characteristic retinal lesions corresponding to the stage of the disease are identified. These include: bone bodies, which are zones of degenerative damage to receptor cells, narrowing of the lumen of the retinal arteries, pallor of the optic nerve head.
Electrophysiological studies carried out to clarify the diagnosis evaluate retinal function most objectively. In addition, special techniques are used to study the capabilities of human vision in the dark and the ability to navigate in low light.
When a diagnosis of retinal pigmentary abiotrophy is made or suspected, it is recommended to examine the patient’s close relatives for early detection of pathology. After all, the disease is hereditary.
Treatment
There are currently no effective treatments for retinitis pigmentosa. In some European countries there is no treatment for this disease. To slow down the rate of progression of disease symptoms, regular courses of conservative therapy are carried out 1-2 times a year. Vasodilators, neurotrophic, vitamin preparations, antioxidants, nootropics, angioprotectors, some biogenic stimulants and biopeptides are used. Physiotherapeutic methods of treatment are used - ultrasound, electro- and phonophoresis, microwave therapy.
Symptoms of retinal abiotrophy
Retinal pigment abiotrophy is characterized by:
- night blindness. It is commonly called hemeralopia. The reason for its appearance is the defeat of the sticks. If the lighting becomes worse, it becomes very difficult for such patients to navigate the surrounding space. Hemeralopia is the first alarming symptom, which should cause alertness among doctors and encourage them to further examine patients with a correct diagnosis;
- changes in vision in the periphery. Its boundaries begin to narrow as the disease progresses from the peripheral areas of the retina to its central part. Over time, “tunnel” or “tubular” vision appears, with a small island of visibility in the center.
Later stages of the disease are characterized by the fact that both visual acuity and color perception in patients decrease, since the cones of the retina are involved in the pathological process. If abiotrophy progresses, blindness, unfortunately, is inevitable.
Treatment of retinal abiotrophy
There are no specific treatment methods for this disease yet, but there are methods that help stop abiotrophy. Treatment is prescribed in the form of vitamins and medications that improve trophism and blood supply to all areas of the retina. These are injections or drops:
- emoxipine;
- taufona;
- mildronate;
- retinalamin.
Recently, Russian specialists began developing a new drug called Alloplant. In addition, patients can be prescribed physical therapy treatment to improve blood flow in the eyeball area. At home, ophthalmologists recommend using Sidorenko glasses, which act in several therapeutic directions at once.
Clinical picture
Retinal pigmentary abiotrophy has a variety of symptoms, since there can be many variants of the mutation that serve as the main cause of the disease. But, if we consider different variants of dystrophy that belong to the same group according to localization, we can identify a number of similar signs.
The peripheral form of the disease begins with damage to the rods, which is why the symptoms begin with hemeralopia. In the case of progression and massive dysfunction of the rods, a person experiences a noticeable deterioration in night vision, which can lead to total loss - Nyctopathia.
Peripheral vision also suffers, as a concentric scotoma develops, against the background of a narrowing of the visual field, which becomes “tubular”. During white spot retinal abiotrophy, severe complications, as a rule, do not occur; daytime vision, as well as color perception, remain normal. Only rarely can changes in the cones be detected, which ultimately cause a decrease in daytime vision (there is a high risk of developing blindness). The course of the pathological process can last for decades, but rapid forms cannot be ruled out.
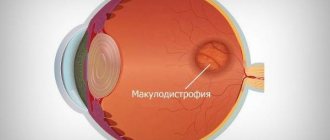
Central abiotrophy negatively affects the cones, which are located in large numbers near the macula, which is why the disease is called macular degeneration. First of all, a person complains of a rapid deterioration in general vision and problems with color discrimination. If all photoreceptors are destroyed, symptoms of central scotoma appear. Without affecting the outermost parts of the retina, peripheral and night vision remains almost unchanged.
There are special types of abiotrophy that cause focal lesions of the photoreceptors of the eye. After them, multiple blind spots appear in the field of view. In severe cases, optic nerve atrophy may develop, resulting in blindness.
Symptoms and diagnosis
- "Night blindness" (hemeralopia). Occurs as a result of damage to the rods of the retinal layer of the eye. With this symptom, patients with abiotrophy have poor orientation in the evening and at night. This sign is one of the early manifestations of pathology.
- Rapid decrease in peripheral vision. This defect is associated with deformation of the cylindrical photosensitive receptors of the inner shell of the eye in the direction from the periphery to the central part. The narrowing of the field of view occurs gradually. Ultimately, a moment comes when patients look at the surrounding space as if into a pipe (tunnel). Hence this phenomenon is called “tunnel vision”.
- Reduced acuity of color and black and white perception. This happens in the later stages of the disease as a result of damage to the second type of photoreceptors, which have a canonical shape, in the center of the retina of the eye. In the absence of proper and timely medical intervention and a sharp progression of abiotrophy, there is a possibility of the patient becoming completely blind.
- Rapid eye fatigue. During the course of the disease, rapid fatigue of the patient's vision organ occurs.
- Other symptoms.
Most of the listed symptoms of pathology often appear more likely at the age of 20–30 years.
Ophthalmologist and geneticist
- these are the two main specialists who should examine and examine people with such a disease of the retinal part of the visual analyzer. When diagnosing abiotrophy of the retina of the visual analyzer, the ophthalmologist analyzes indicators such as visual acuity and parameters of the patient’s visual field. The fundus of the eye is carefully checked, where specific changes in the retina are detected.
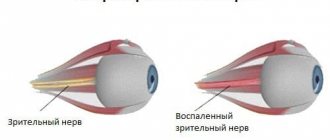
On the inner surface of the eyeball, one can clearly see the narrowing of the blood vessels that supply blood to the retinal layer of the visual analyzer, and the deformation of the color of the disc on the optic nerve. Electroretinography is performed, with the help of which the functionality of the reticular inner layer of the organ of vision is assessed.
To diagnose the presence of “night blindness” syndrome, the patient’s condition is examined using special techniques, when a person’s orientation in a darkened space and dark adaptation are assessed.
If there is a suspicion of heredity of abiotrophy, it is advisable to study the anamnesis of the patient’s direct relatives with a geneticist for the early detection of mutations in genes characteristic of this disease.
Retinal pigmentary degeneration
The disease occurs in childhood or adolescence and progresses slowly. Initially, there are complaints of decreased vision at dusk and difficulty in orientation. Often, hemeralopia is the first sign of the process and can persist for several years before changes appear that can be detected ophthalmoscopically.
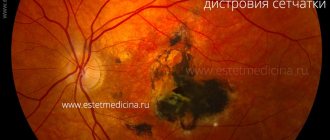
Later, characteristic pigment foci appear on the periphery of the fundus, shaped like bone bodies. Gradually their number increases and they spread towards the center. The retinal vessels narrow sharply. Certain areas of the retina become discolored, and choroidal vessels can be seen in these places. The optic disc becomes atrophic and takes on a yellowish-white, waxy color (waxy atrophy). Visual acuity remains high for a long time, the field of vision gradually narrows concentrically, and a characteristic annular scotoma appears. As the process progresses, the field of view narrows more and more and becomes tubular. In advanced stages, posterior polar cataracts, secondary glaucoma and vitreous opacities often develop. Visual acuity decreases sharply. Sometimes retinal pigmentary degeneration occurs in an atypical manner.
A non-pigmented form of pigmentary degeneration. In this form, waxy atrophy of the optic nerve, narrowing of retinal vessels, hemeralopia and characteristic changes in the visual field are detected, but there are no pigment deposits in the fundus.
Unilateral pigmentary degeneration is a very rare type of disease. The clinical picture of the affected eye is the same as with bilateral retinal degeneration. Glaucoma is also often found in the affected eye.
Whitish dotted retinitis is characterized by the appearance on the fundus of the eye, excluding the very central parts, of numerous, in most cases small, rarely larger, white round, sharply demarcated spots. The disease has two forms: stationary and progressive. With a progressive form, the retinal vessels gradually become narrow, optic nerve atrophy develops, and pigment deposits appear. In these cases, along with hemeralopia, the field of vision sharply narrows and visual acuity decreases.
Central pigmentary degeneration of the retina is characterized by hemeralopia, the development of paracentral and central scotoma, deposition of pigment in the form of “bone bodies” and lumps in the macular and paramacular areas.
The paravenous form of retinal pigmentary degeneration is a disease in which coarse, dark-colored pigment deposits are grouped along the large retinal veins.
Lobular atrophy of the choroid and retina is very rare. This form is most characterized by the occurrence of hemeralopia. As the process progresses, visual acuity gradually decreases, and the field of vision narrows concentrically. In the fundus of the eye, starting from the periphery, areas of atrophy of the retina and choroid are formed, which, in the form of wide stripes with rounded edges, move closer to the center. Scattered islands of pigment may be found in areas of atrophy.