The term dystrophy is translated from Latin as “tissue nutrition disorder.” The basis of dystrophic processes of any localization is a violation of metabolic processes at the cellular level, which leads first to the appearance of structural changes, and then to functional failure.
Retinal dystrophy is a complex pathology of the retina of the eye, which leads to a persistent deterioration in visual acuity. The reason for its appearance is a complex of factors that can be divided into external (exogenous) and internal (endogenous). The immediate cause of retinal dystrophy is vascular disorders, which trigger a cascade of pathological reactions, ultimately leading to a pronounced decrease in vision. For a long time, the disease remains completely unnoticeable and is often detected only when complications develop.
In the treatment of most eye diseases, the time of their detection is critical, because the earlier adequate treatment is started, the better the prognosis in terms of vision preservation. Don't deprive yourself of the opportunity to see well; get your eyes examined every year, even if you think you have no symptoms of eye disease! A simple, quick, and inexpensive eye exam with an ophthalmologist can help you preserve your vision for years to come!
Causes of retinal dystrophy
Quite often, retinal dystrophies are genetically determined, but there are a number of factors that increase the likelihood of developing the disease. These include:
- elderly age
- diseases of the cardiovascular system (hypertension, atherosclerosis)
- diabetes
- overweight
- bad habits (smoking, alcohol abuse, unhealthy diet)
- moderate and high myopia
- severe infectious diseases, chronic stress
- previous eye inflammations, eyeball injuries
How does retinal dystrophy manifest?
A common symptom for any form of dystrophy is progressive deterioration of vision, as well as impairment of visual fields. When examining the eyes, scotomas (areas of vision loss), distortion of the visible image of objects, deterioration in orientation at dusk and in conditions of limited illumination may be detected. A patient with retinal dystrophy may notice curvature of straight lines, opaque spots in the field of vision, letters falling out while reading, and changes in color perception.
At an early stage, retinal dystrophy may not cause any discomfort. So if you or a loved one is at risk, don't skip your annual eye exam with a qualified ophthalmologist.
At-risk groups
Source: setchatkaglaza.ru
Most often, dystrophy occurs in older people. The risk group includes people suffering from myopia, as well as those who have vascular diseases, diabetes, and hypertension.
The tendency to this disease is inherited, so children with retinal dystrophy are recommended to undergo fundus examinations more often.
According to statistics, this disease is more common in people with white skin and blue irises. Women get sick more often than men.
What are the types of retinal dystrophies?
Retinal dystrophy is a collective concept that unites pathologies of the retina of the eye that are different in nature and manifestations. By their nature, retinal dystrophies are divided into primary, hereditarily caused, and secondary, acquired, caused by general diseases of the body.
Primary retinal dystrophies are a variety of different pathologies that are inherited. They usually appear in childhood and cause persistent progressive vision loss, eventually leading to blindness. Examples of primary retinal dystrophies are Leber's amaurosis, retinal pigmentary dystrophy, Wagner's disease, Best's dystrophy, Stargardt's disease, and white dotted retinal dystrophy.
Secondary (acquired) dystrophies, depending on the predominant area of retinal damage, are divided into central and peripheral.
What is the threat?
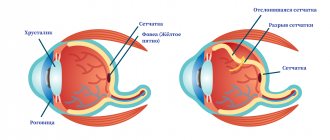
The prognosis directly depends on the type of dystrophy. With progressive myopia (myopia), when the eye lengthens throughout life, degenerative changes develop in the fundus of the eye - the retina overstretches and pathological foci form on it.
They are a possible source of rupture, so patients with moderate and high myopia are at risk for retinal detachment.
Retinitis pigmentosa is a chronic, slowly progressive condition that affects young people. With relatively good central vision, at advanced stages, only a small “window” of healthy retina remains. The patient sees the world as if through a narrow tube.
Important
Minus: there is a family predisposition to this disease. Plus: temporary stabilization and process control are possible with timely maintenance treatment at least 2 times a year.
Best's vitelline dystrophy occurs in three stages: the formation of a cyst, its rupture with hemorrhage and the formation of a scar on the affected area. Accordingly, the final visual functions are determined by the localization of the process.
Stargardt's macular degeneration and Franceschetti's yellow-spotted degeneration lead to a noticeable decrease in vision because they involve the central part of the retina - the macula.
The development of secondary dystrophies is associated with the progression of the underlying disease - usually against the background of chronic hypertension, kidney disease or leukemia, angiopathy develops, which can develop into retinopathy. Its outcome is dystrophy.
Speaking about retinal dystrophy in older people, it is necessary to clarify that some types of peripheral changes are more dangerous in terms of possible detachment than others. For example, retinoschisis (separation) is much less likely to develop into a rupture than the same lattice dystrophy.
As for age-related macular degeneration, it certainly reduces the patient’s quality of vision, but in this case, the level of discomfort depends on the form - dry or exudative. The first is usually less aggressive, the second requires active intervention and significantly affects the functions of the eye.
Central retinal dystrophies
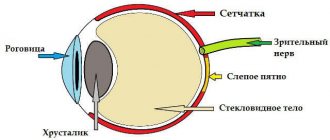
Since this form of retinal dystrophy affects the area of the macula (macula, the central fovea of the retina), the disease is also called macular degeneration. The macula, or macula, is the area of the retina responsible for the best visual acuity and the distinction of fine details in the image. When this area is affected, central vision is affected, but peripheral vision remains unchanged. It is difficult for a person with central retinal dystrophy to read, drive a car, draw, write, embroider, etc.
At a young age, central retinal dystrophy is often detected with myopia; in old age, age-related macular degeneration is diagnosed. Timely detection and regular therapy can stop the progression of disease symptoms and preserve vision for many years.
Peripheral retinal dystrophies
In this case, changes are detected in the periphery of the retina, that is, in those areas that do not participate in the formation of clear vision. Peripheral retinal dystrophies, as a rule, are not accompanied by a clear deterioration in vision, but increase the risk of developing severe eye pathologies, such as retinal detachment.
Peripheral dystrophies of varying severity are diagnosed in the majority of myopic people. Therefore, if you wear glasses or contacts, be sure to undergo an annual fundus examination by an ophthalmologist. If the doctor finds areas of thinning or tears in the retina, you will be offered restrictive laser photocoagulation of the retina.
Best's disease and adult vitelliform dystrophy
- Genetic research
- Histological and histochemical studies
- Classification
- Clinical manifestations
- Diagnostics
- Differential diagnosis
- Treatment
Best's disease is a rare bilateral macular degeneration with a variety of asymmetric clinical manifestations, inherited in an autosomal dominant manner. The disease was first described back in 1883 by JE Adams as “peculiar” changes in the macula.
In 1905, F. Best reported 8 patients with macular degeneration from a large family of 59 people, and suggested that the disease was transmitted in an autosomal dominant manner.
The disease is known in the literature under the following names:
- Best's vitelliform dystrophy,
- central exudative retinal detachment,
- hereditary macular pseudocysts,
- hereditary vitelliform macular degeneration
The most characteristic manifestation of Best's disease is changes in the macula, resembling egg yolk, with a diameter of 0.3 to 3 RD.
Adult vitelliform macular degeneration was first described by JDM Gass in 1974. The disease develops at the age of 40-50 years and is characterized by bilateral symmetrical local subretinal changes in the fovea. The lesions are usually round in shape and yellow in color, their diameter varies from 0.3 to 0.5 RD. Visual impairment is minimal.
Unlike Best's disease, foveal changes in adult vitelliform macular degeneration develop in adulthood, are smaller in size and do not progress.
Genetic research
Best's disease is a disease with an autosomal dominant mode of inheritance and high penetrance. The gene responsible for the development of vitelliform macular degeneration, which is called “bestrophin,” is localized on the long arm of chromosome 11 in the interval between the D11S903 and PYGM loci (the gene encoding muscle glycogen phosphorylase) or, as later studies showed, between the D11S986h D11S480 markers.
In addition, the ROM1 gene, encoding a photoreceptor-specific membrane protein, was mapped to this region. In this regard, it has been suggested that Best's disease is the result of mutations occurring in the ROM1 gene.
N. Stohr and V.N. Weber (1995), mapping the ROM1 gene as well as the D11S986, UGB (uteroglobin gene) and PYGM loci, showed that UGB is localized proximal to ROM1 and both genes recombine in Best disease. The authors concluded that the ROM1 gene is not related to Best's disease.
Overlapping approximately 40 cM, the region 11q13-14, in which the bestrophin gene is localized, is notorious to ophthalmogeneticists as the “refuge” of many genes responsible for the development of a number of retinal diseases, in particular the oculocutaneous form of albinism, Usher syndrome (“myosin 7A”), Bardet-Biedl, an autosomal dominant form of familial exudative vitreoretinopathy.
RE Ferrell et al. (1983), when examining patients with atypical familial vitelliform macular degeneration, revealed its connection with the classical marker GPT1, localized on the long arm of chromosome 8. Some patients from the family they examined had a normal EOG, and the diameter of the foci of vitelliform changes in the macula in all cases did not exceed 1 RD.
The high variability in the clinical manifestations of Best's disease is largely due to genetic heterogeneity and, in addition, may be due to hitherto unknown mutations. S. Nordstrom and W. Thorburn (1980) reported a family in which a homozygous father had 11 daughters with various clinical manifestations of Best's disease.
Adult vitelliform macular degeneration
Although the literature on adult vitelliform dystrophy does not provide data on familial cases, it is believed that the disease is transmitted in an autosomal dominant manner. Recently, there have been reports that patients with adult vitelliform dystrophy have the Y258Stop mutation in the peripherin/LAH gene. In a study of the coding region of the RDS gene in 18% of 28 patients with adult vitelliform macular degeneration, the authors identified five new mutations, two of which were neutral or polymorphisms. Thus, it cannot be ruled out that in some cases changes in the RDS gene also lead to the development of vitelliform dystrophy, but the mechanism by which specific mutations in this gene can cause retinal dystrophies, including macular degeneration, is still unknown.
Histological and histochemical studies
Studies using light and electron microscopy have shown that in the early stages of Best's disease, pigment epithelial cells are engulfed by cytosomes containing an abnormal, unidentifiable substance. Between the cells of the retinal pigment epithelium and the neuroepithelium, granules of a lipofuscin-like substance accumulate, the integrity of Bruch's membrane and the structure of the neural elements of the retina are disrupted: most of the outer segments of the photoreceptors disappear or degenerate, while acidic mucopolysaccharides accumulate in the inner segments of the photoreceptors of the affected areas of the retina. Capillaries penetrate into the subretinal space through the altered Bruch's membrane.
In stage II, a significant amount of granules of a substance histochemically close to lipofuscin accumulates inside the cells of the retinal pigment epithelium in the macula, as well as macrophages in the subretinal space and in the choroid. In the later stages of the disease, flattening of the retinal pigment epithelial cells in the macula is observed, the diameter of which exceeds the norm by 1.5-2 times.
In patients with vitelliform dystrophy of older and older age, diffuse deposits of lipofuscin-like substance were detected in the pigment epithelium layer, in the internal segments of photoreceptors, inside Müller cells and even in the vitreous body.
Classification
Depending on the ophthalmoscopic manifestations, five stages are distinguished in the course of Best's disease:
- stage O - there is no change in the macula, but an abnormal EOG is recorded
- stage I - minimal pigment disturbances in the macula, hypopigmentation;
- stage II - classic vitelliform cyst in the macula;
- stage III - rupture of the cyst and various phases of resorption of its contents;
- stage IV - resorption of the cyst contents, formation of a fibroglial scar with or without subretinal neovascularization.
At the same time, there is no convincing evidence that in most patients with Best's disease the evolution of macular changes occurs sequentially through all stages.
Clinical manifestations
The course of Best's disease is usually asymptomatic; it is detected accidentally during examination of children aged 2-6 years. Ophthalmoscopic changes are asymmetrical in most cases. At stage zero, the fundus of the eye in children usually looks normal. Sometimes there is a weakening of the foveal reflex. Adults with a similar ophthalmoscopic picture are regarded as carriers of a pathological gene, which is confirmed by a decrease in the Arden coefficient - the ratio of the light peak to the dark decline in the EOG.
- Stage I , or “previtelliform,” stage of Best’s disease is characterized by the appearance of small yellow spots in the macula.
- In stage II of the disease, the size of vitelliform cysts, resembling “egg yolk”, can reach the diameter of the disc. The changes are often bilateral, but rarely symmetrical. Visual acuity at this stage does not correspond to ophthalmoscopic manifestations, remaining even in elderly people in the range from 0.4 to 0.9.
- A decrease in visual acuity is noted in stage III of the disease, when individual vitelliform cysts rupture, causing an association with “scrambled eggs.”
- Later, as a result of partial resorption and displacement of the lipofuscin-like contents of the cyst, a picture of pseudohypopyon is formed.
At any stage of the disease, patients with Best's disease may experience subretinal hemorrhages. In approximately 5% of cases, a subretinal neovascular membrane forms.
SA Miller et al. (1976) reported a 9-year-old boy with Best's disease who was found to have an intact vitelliform cyst in the right eye and a ruptured, partially resorbed cyst with subretinal hemorrhages and a neovascular membrane in the left eye.
A.R. Schachat et al. (1985) observed a casuistic case of the development of a macular hole and rhegmatogenous retinal detachment in a patient with Best's disease. In old age, patients with this pathology often develop choroidal sclerosis in the macular region due to the loss of choriocapillaris and atrophy of the retinal pigment epithelium.
There are reports in the literature of multiple vitelliform lesions in Best disease. In these cases, macular vitelliform changes are combined with extrafoveal retinal lesions, usually localized along the superotemporal vascular arcade. There are usually 2 to 5 extramacular vitelliform lesions. Their diameter is slightly smaller than the size of macular changes and does not exceed 0.2-1 RD.
Satellite lesions may evolve asynchronously with respect to each other and changes in the macula, or may be at the same stage of development.
Diagnostics
Fluorescein angiography
The angiographic picture of Best's disease varies depending on the stage of the disease. In the initial stage of the disease, there is a lack of fluorescence in the area of the cyst (the so-called fluorescence block). In the previtelliform stage of the disease, small fenestrated defects caused by local hyperfluorescence are detected in areas of atrophy of the pigment epithelium in the macula.
After the cyst ruptures (the “pseudohypopyon” stage), hyperfluorescence is detected in its upper half, while a “block” of fluorescence remains in the lower half. In the late stage of the disease, when the lipofuscin-like contents of the cyst are partially resorbed, fenestrated defects are detected in the macula.
Psychophysical research
- Complaints. Patients with Best's disease typically complain of blurred vision, decreased visual acuity, difficulty reading small print, and metamorphopsia.
- Visual acuity. Visual acuity in patients with Best's disease varies depending on the stage of the disease from 0.02 to 1.0. During long-term observation of 58 patients with Best's disease in various stages, R. Clement (1991) found that 43% of them had visual acuity of 0.5 or higher. G. A. Fishman et al. (1993), having examined 47 patients with Best's disease stages II-IV aged 5-72 years from 23 families, found that 41% of them had visual acuity of at least 0.3. Meanwhile, the authors noted that only 20% of patients over 40 years of age had vision that met the requirements of the driver’s commission, and in all patients over 50 years of age with ophthalmoscopic changes corresponding to stages III-IV of the disease, visual acuity of the better-seeing eye did not exceed 0.1-0 ,3.
- Color vision. Color perception is normal in most patients.
- Line of sight. The peripheral boundaries of the visual field remain normal. Computer static perimetry may reveal a relative central scotoma in some patients. The threshold of brightness sensitivity in the initial stage of the disease in patients with high visual acuity does not change.
- Electrophysiological studies. General and local ERG remain normal in Best's disease. The only reliable test to diagnose Best's disease is electrooculography. Characteristic signs of Best's disease are low baseline potential and a decrease in the Arden ratio - the ratio of the light peak to the dark trough. The Arden coefficient in patients with Best disease does not exceed 1.5 (150%). EOG changes are recorded not only in patients with clinical manifestations of the disease, but also in carriers of the pathological gene. In patients with adult vitelliform macular degeneration, the EOG is usually unchanged.
Differential diagnosis
An accurate diagnosis in patients with Best's disease and adult vitelliform dystrophy is established based on the ophthalmoscopic picture and the results of ERG and EOG recording. In difficult cases, examination of other family members can help in diagnosis.
Best's disease must be differentiated from pigment epithelial detachment, Coats' disease, and acute toxoplasmosis chorioretinitis.
- Detachment of the pigment epithelium. Difficulties in diagnosis arise in patients with an atypical clinical picture and exudative changes in the macula. The disease is often unilateral. The general ERG does not change, the local ERG may be reduced. EOG is normal in most cases. Fluorescein angiography in the late phase in patients with retinal pigment epithelial detachment reveals hyperfluorescence.
- Coats disease. In most patients with Coats' retinitis, during the first examination by an ophthalmologist, a prominent focus of yellow hard exudate, reminiscent of vitelliform changes in Best's disease, is detected in the macula. Exudative changes in the macula are often combined with subretinal hemorrhages and neovascularization. The diagnosis is based on the results of ophthalmoscopy: when examining the periphery of the fundus using a head-mounted ophthalmoscope and lenses of 20-30 diopters or biomicroscopy with a three-mirror Goldmann lens, vascular anomalies (telangiectasia, micro- and macroaneurysms, arteriovenous shunts), more often localized, are detected in all patients with Coats disease in the temporal half of the retina. The disease is sporadic. In 92-95% of patients, one eye is affected. The EOG does not change in the initial and advanced (PA) stages of the disease. Visual acuity in patients with Coats retinitis in the presence of exudative changes in the macula does not exceed 0.4.
- Acute toxoplasmosis chorioretinitis. Diagnostic difficulties usually arise when examining patients with an acquired form of toxoplasmosis and exudative-hemorrhagic changes in the macula. The disease is usually unilateral. Choroiretinal lesions are combined with vitreitis of varying severity, and sometimes with changes in the anterior segment. Such patients usually complain of a sudden significant decrease in visual acuity, which in most cases varies from 0.01 to 0.2. With static perimetry, an increase in the brightness sensitivity threshold and an absolute or relative central scotoma are detected. The diagnosis of toxoplasmosis can be confirmed by positive results of immunological studies.
Treatment
There is no effective treatment for patients with Best's disease and adult vitelliform macular degeneration, and patients should undergo a comprehensive eye examination, including dilation, once or twice a year to rule out any possible complications such as CNV, macular holes, or retinal detachments. If vision is impaired, patients should be referred for vision loss testing and medical rehabilitation.
Intravitreal injections of ranibizumab may be effective in the short term. The prognosis for vision is fairly good because the disease usually results in slowly progressive vision loss, and most patients maintain good vision in at least one eye until the late stages of the disease. Vitelliform degeneration usually disappears during life, but in the later atrophic stages of the disease, vision loss, the formation of macular holes, or retinal detachment may occur.
Maintenance therapy:
- Mildronate 5.0 IV No. 10, then 1 capsule 2 times a day, 1 month
- Mexidol 2.0 IM No. 10, then 1 tablet. 3 times a day, 1 month
- Emoxipin 1% 1 drop 3 times a day, 1 month
- Milgamma 2.0 No. 5 w/m h/d
- Nutrof forte 1 tablet 1 time per day continuously
Treatment of retinal dystrophy
Timely treatment of retinal dystrophy helps improve visual acuity, stop the progression of pathological changes in the tissues of the eye and prevent the occurrence of serious complications.
An integrated approach is used to treat retinal dystrophies. Due to the fact that most cases of the disease are hereditary, treatment of the disease is symptomatic, aimed at maintaining sufficient visual acuity.
Drug treatment includes the use of drugs with different mechanisms of action, the purpose of which is to improve the nutrition of retinal tissue. Typically, vasodilators (no-spa, papaverine), antiplatelet agents (aspirin, clopidogrel), complex vitamin preparations that strengthen the walls of retinal vessels (ascorutin, preparations with lutein), biogenic stimulants and peptides (retinalamine, aloe extract) are used. If an exudative form of AMD is detected, intravitreal injections of drugs from the group of neoangiogenesis blockers (anti-VEGF drugs), dexamethasone, and absorbable drugs (heparin, etamsylate) are indicated.
Physiotherapeutic treatment for retinal dystrophy includes magnetic therapy, laser stimulation of the retina, electrophoresis with drugs that improve tissue nutrition (no-spa, nicotinic acid, etc.).
Hardware treatment is also possible at home - using the AMBO-01 device, designed specifically for use in patients with pathologies of the retina and optic nerve.
The most effective treatment method for patients with retinal dystrophies is laser coagulation (photocoagulation) of the retina. The goal of such treatment is to isolate the altered areas of the retina from the surrounding healthy tissue to prevent the progression of the disease and the development of possible complications. Local exposure to laser beams increases the temperature in the treatment area and causes limited tissue coagulation and the formation of strong adhesions between the retina and the adjacent choroid.
Surgical treatment is indicated for the detection of rough adhesions (shvart) between the retina and the vitreous body of the eye, as well as neovascular membranes. For this purpose, vitreoretinal operations are used (vitrectomy, scleroplasty, extrascleral filling, etc.).
Diagnostic methods
Source: visus-1.ru
In addition to a basic examination for retinal diseases, the following studies must be carried out to make a correct diagnosis:
- Optical coherence tomography of the retina is a non-contact, high-precision study of the retina at the level of tissue structure.
The method allows you to obtain a comprehensive picture of the ultrastructure of any part of the retina.
All measurements are carried out in microns and very accurately reflect the slightest changes. Optical coherence tomography is one of the most accurate and universal research methods in ophthalmology, absolutely necessary for retinal diseases.
RTVue-100 can significantly expand research capabilities and increase diagnostic accuracy, and is absolutely necessary in the diagnosis and treatment of retinal diseases.
In modern ophthalmology, it is the results of this study that are decisive in choosing treatment tactics and monitoring its effectiveness.
- Quantitative static perimetry is a very accurate method for studying the visual field
Allows you to identify the slightest functional defects in the visual field and obtain their quantitative assessment. Control studies throughout the year make it possible to objectively assess changes in the size and shape of the pathological focus.
Comparison of images obtained over time makes it possible to assess the dynamics of the disease and the effectiveness of the treatment.
- Fundus photography and phage (fluorescein angiography)
Quite often, during an examination, the doctor needs not only to examine in detail the picture of the fundus (retina, eye vessels, optic nerve head), but also to obtain an enlarged image of certain details, and also to record existing changes.
This is especially important in cases where further monitoring of the process is expected in order to assess the effectiveness of treatment and monitor its dynamics. In these cases, the doctor resorts to special photography of the fundus.
Such photography is carried out using a digital fundus camera. The resulting images are stored in an archive; the image is also copied on a CD and given to the patient.
- Fluorescein angiography of the eye (FA) is an objective method for studying the vessels of the eye when they are contrasted with fluorescein.
The method is indispensable for clarifying the diagnosis of retinal diseases. Without this study, it is impossible to make an informed decision about the treatment strategy for a particular patient with retinal disease.
PAH is necessary for the following diseases:
- age-related macular degeneration,
- diabetic retinopathy and diabetic macular edema,
- retinal vein thrombosis,
- central serous chorioretinopathy,
- inflammatory processes in the retina and optic nerve.
Diagnosis of the disease includes examination of the fundus, determination of visual acuity, assessment of visual fields and assessment of color perception. It is more difficult to examine the fundus if peripheral dystrophy is suspected.
It is necessary to dilate the pupil with medication and examine the fundus using a three-mirror Goldmann lens.
Using optical coherence tomography, the doctor can take a three-dimensional picture of the retina of the eye. The viability of retinal cells and nerve endings is studied using electrophysiological studies. Sometimes ultrasound is used.
In order to confirm or refute the diagnosis of retinal dystrophy, it is necessary to undergo a thorough examination of the visual system.
Examination of patients with suspected retinal dystrophy includes:
- determination of visual acuity;
- study of visual fields (perimetry) in order to assess the condition of the retina in its periphery;
- optical coherence tomography;
- electrophysiological study - determination of the viability of nerve cells of the retina and optic nerve;
- ultrasound examination of the internal structures of the eye - A-scan, B-scan;
- measurement of intraocular pressure (tonometry);
- fundus examination (ophthalmoscopy).
Other instrumental methods:
- visometry – subjective determination of visual acuity, primary selection of correction (glasses);
- perimetry – analysis of visual fields, search for those “blind” spots;
- optical coherence tomography is a modern method of non-contact diagnostics, which consists of step-by-step imaging of the retina.