Deterioration of vision in the dark is the very first sign of the disease - it may be the only symptom for many weeks or even years.
A person does not adapt well when moving from light to darkness. Further, blindness develops in the dark, while daytime perception is fine.
As the disease progresses, the outlines of objects become distorted, and blind spots appear in the field of vision. As a result, the field of vision narrows towards the center and the sick person sees everything as if through a pipe, and ceases to distinguish shades of blue.
Share
Tweet
Share
Cool
Send
General information about the disease
Retinal pigmentary degeneration has a low prevalence, on average affecting 1 in 5 thousand people. According to statistics, the disease more often affects men, although it is not gender specific.
Pathology is detected at the age of 20-30 years. Visual acuity decreases by 45-50 years and reaches 0.1.
Modern medicine is not able to completely overcome the disease and treatment comes down to the struggle to maintain visual acuity at an advanced stage.
Retinitis pigmentosa is a genetic heterogeneous disease that appears as a result of disruption of the functioning of the retinal pigment epithelium with the development of other disorders.
The manifestation and severity of symptoms depend on the form of the disease. Pathology can manifest itself both at an early age and in adulthood and is entirely dependent on gene mutations.
Reference! Retinitis pigmentosa is inherited along any parental line. Children born in consanguineous marriages are at risk.
ICD-10 code
H35.5 Hereditary retinal dystrophies.
↑ Retinitis pigmentosa
↑ SYNONYMS
Tapetoretinal dystrophy, rod-cone dystrophy, primary retinal pigmentary dystrophy.
↑ DEFINITION
Retinitis pigmentosa
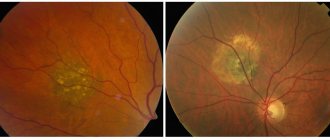
- a hereditary disease that develops in early childhood, characterized by dystrophic changes in the pigment epithelium and photoreceptors and having a typical picture of the fundus with pigmented foci similar to pigmented bone bodies.
↑ ICD-10 CODE
H35.5 Hereditary retinal dystrophies.
↑ EPIDEMIOLOGY
Retinitis pigmentosa
- the most common hereditary disease of the retina: the incidence rate in the population is 1 in 3 thousand people, there are about 1.5 million patients worldwide. The most common form is autosomal dominant (from 9 to 39% of patients in Switzerland and the UK); more rare - autosomal recessive (from 9 to 15% in Switzerland and the UK); the most common type of disease is X-linked (from 1 to 15% in Switzerland, Russia and the UK). The prevalence of retinitis pigmentosa in Birmingham (England) is 1:5000 in all age groups; in Norway - 1:4400, the frequency of sporadic cases is 1:3700, in some ethnic groups - 1:1800.
↑ CLASSIFICATION
Genetic types:
autosomal dominant, autosomal recessive, X-linked.
Clinical forms:
typical (retinitis pigmentosa): atypical (pigmentless form of retinitis pigmentosa, sectoral horseshoe-shaped retinitis pigmentosa, inverted (central form); white-spot retinitis pigmentosa (retinitis punctata albescens).
↑ ETIOLOGY AND PATHOGENESIS
Molecular defects cause differences in pathogenetic mechanisms and cause the development of retinitis pigmentosa with different types of inheritance. 120 genes associated with retinitis pigmentosa have been cloned, of which 52 have been mapped. Each form of the disease is caused by one or more genes expressed in photoreceptor cells and pigment epithelium. Most mutations leading to retinal degeneration occur in genes encoding the synthesis of proteins of the phototransduction cascade and retinol acetate (vitamin A) metabolism (rhodopsin, alpha and beta subunits of cyclic guanosine monophosphate, arrestin, ATP-binding cassette transporter retinal, G protein, cellular retinaldehyde binding protein and the RPGR transduction gene, etc.), as well as in the genes responsible for the transcription factor.
There are allelic and non-allelic varieties of retinitis pigmentosa.
Risk factors for developing retinitis pigmentosa
- consanguineous marriages, as well as the presence of diagnosed cases of retinitis pigmentosa in the family.
↑ CLINICAL PICTURE
Characterized by a triad of symptoms:
typical pigment foci (“bone bodies” on the middle periphery of the fundus and along the venules): waxy pallor of the optic disc, in some cases with perepapillary gliosis; atrophy of the retinal pigment epithelium with white spots at the level of the pigment epithelium layer, narrowing of arterioles. Patients with retinitis pigmentosa may develop pigmentary changes in the macular area over time due to degeneration of photoreceptors. Involvement of the macula is accompanied by decreased visual acuity and posterior vitreous detachment with pigment deposition in it.
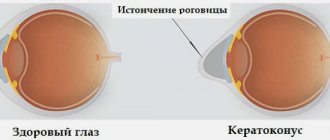
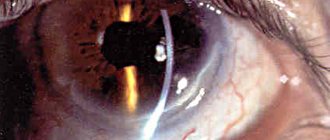
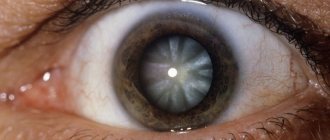
Macular edema may occur due to the penetration of fluid from the choroid through the pigment epithelium, and as the process develops, preretinal macular fibrosis. In patients with retinitis pigmentosa, optic disc drusen, posterior subcapsular cataract, open-angle glaucoma, keratoconus and myopia are found more often, with a higher frequency than in the general population. The choroid remains intact for a long time and is involved in the process only in the later stages of the disease.
Due to damage to the rod system, night blindness occurs (lack of night vision, nyctalopia). Dark adaptation is impaired already in the initial stage of the disease, the threshold of light sensitivity is increased in both the rod and cone parts.
Other forms of retinitis pigmentosa
Other forms of retinitis pigmentosa include: inverted retinitis pigmentosa, retinitis pigmentosa without pigment, retinitis punctata albescens and pseudopigmentary retinitis. Each of these forms has a characteristic ophthalmoscopic picture and ERG symptoms.
Retinitis pigmentosa inverted (central form).
Unlike retinitis pigmentosa, the disease begins in the macular region, and the cone system is affected more significantly than the rod system. First of all, central and color vision decrease, photophobia (photophobia) appears.
In the macular area, characteristic pigment changes are found, which can be combined with dystrophic changes in the periphery. Then, along with hemeralopia* (lack of daytime vision), characteristic of the pathology of the cone system, a decrease in twilight and night vision, nyctalopia, is noted.
* The term "hemeralopia" was previously erroneously used to refer to impaired twilight vision.
A central scotoma is determined in the field of view; on the ERG, the cone components are significantly reduced compared to the rod components.
Retinitis pigmentosa without pigment.
The name is associated with the absence of pigment deposits in the form of bone bodies characteristic of retinitis pigmentosa with symptoms similar to retinitis pigmentosa and unrecorded ERG.
White spots retinitis pigmentosa (retinitis punctata albescens).
A characteristic ophthalmoscopic sign is multiple white dotted spots throughout the fundus of the eye with or without accompanying pigmentary changes (“moth-eaten tissue”). Functional symptoms are similar to retinitis pigmentosa. Differential diagnosis is necessary with stationary congenital night blindness and fundus albipunctatus albipunctatus.
↑ DIAGNOSTICS
Anamnesis
Information about cases of the disease in relatives is important.
Laboratory research
Histologically, in retinitis pigmentosa, dystrophic changes are found mainly in the outer segments of photoreceptors, atrophy and hypertrophy of the pigment epithelium, its flattening and disorganization. An increase or decrease in the amount of lipofuscin, melanin and phagosomes is detected in the cells. In the retina, migration of pigment in the form of melanin granules is observed.
Instrumental studies
Using functional research methods, progressive changes in photoreceptors are detected.
With perimetry in the middle periphery (30-50°), complete and incomplete annular scotomas are noticeable, expanding to the periphery and center as the disease progresses. In the late stage, the field of vision narrows concentrically to 10°, only the central field of vision is preserved (tubular, or tunnel vision).
The absence or sharp decrease in the amplitude of general ERG components is a pathognomonic sign of retinitis pigmentosa.
Local ERG remains normal for a long time, and changes occur when the cone system of the macular region is involved in the pathological process. In carriers of the pathological gene, a reduced ERG and an extension of the latent period of the ERG b-wave are noted, despite a normal ophthalmoscopic picture of the fundus.
When studying tempo adaptation, an increase in the rod threshold is noted.
Early fluorescence angiography can detect defects away from the macula associated with atrophy of the pigment epithelium; cystic changes with an uneven surface and cystoid macular edema.
Differential diagnosis
Differential diagnosis is carried out with pseudopigmentary retinitis, which can be caused by infections (rubella, syphilis): intoxications caused by taking medications (chloroquine, phenothiazine derivatives, etc.); injuries; other hereditary or acquired diseases of the retina (geographic atrophy, reticular degeneration, sarcoidosis, birdhot retinochoroiditis, crystallin dystrophy, Fogg-Koyanagi-Harada disease, Leber congenital amaurosis, retinitis albipunctatus, etc.).
A heterogeneous group of diseases with various non-hereditary etiologies, in which changes occur in the fundus of the eye, similar to those in retinitis pigmentosa. The development of pseudopigmentary retinitis is promoted by inflammatory processes in the retina and choroid, side effects of drugs (thioridazine, chloroquine, deferoxamine, clofazimine, etc.), conditions after injury and retinal detachment, etc.
The leading differential symptom is normal or slightly decreased ERG. With this form of retinitis, there is never an unrecorded or sharply reduced ERG.
An example of a diagnosis formulation
Retinitis pigmentosa, retinitis pigmentosa pigmentosa, retinitis pigmentosa inverted (central form), retinitis pigmentosa without pigment.
↑ TREATMENT
Treatment Goals
There is no pathogenetically based treatment for retinitis pigmentosa, which is due to the hereditary nature of the disease and dystrophic and degenerative changes in the retina that cause remodeling processes, preventing the transmission of information to the central parts of the visual system.
Further management
• Monitoring of visual functions, ERG registration.
• FA for suspected macular edema.
↑ INFORMATION FOR THE PATIENT
It is recommended to use dark safety glasses that do not transmit short-wave radiation to prevent the damaging effects of light, correct spectacle correction of visual acuity, and possibly symptomatic treatment.
Studying in specialized schools for the visually impaired, choosing a profession according to the capabilities of the visual and other systems, wearing light filters. Systematic observation by specialists for timely correction of visual acuity, treatment of macular edema and cataracts.
Causes
Any form of retinitis pigmentosa can be caused by one or more genes. Many mutations in the rhodopsin and peripherin genes cause a range of phenotypic manifestations of the disease.
The origin of retinitis pigmentosa is associated with metabolic disorders in photoreceptors and pigment epithelium. The brain stops receiving the necessary signals and this leads to the accumulation of toxic substances in the retina.
Molecular mutations in genes disrupt the production of rhodopsin (a precursor to retinol), which is vital for normal eye function.
Development ways:
- Gender dependence - transmitted from mother to son along with the X chromosome.
- Autosomal recessive - transmitted from two parents at once.
- Autosomal dominant - passed from one parent.
- Not hereditary - very rare. Usually the disease is passed on from generation to generation.
Associated terms
RP may be: (1) non-syndromic, that is, it occurs alone, without any other clinical findings, (2) Syndromic with other neurosensory disorders, developmental disorders, or complex clinical findings, or (3) Secondary to other systemic diseases [4].
- RP combined with deafness (congenital or progressive) is called Usher syndrome.
- RP in combination with ophthalmoplegia, dysphagia, ataxia, and heart defects seen as a mitochondrial DNA disorder - Bardet-Biedl syndrome (also known as ragged red fiber myopathy)
- RP in combination with retardation, peripheral neuropathy, acanthotic (spiked) red blood cells, ataxia, steatorrhea, indicates VLDL deficiency, apparently due to abetalipoproteinemia.
- RP with a clinically visible association with a number of other rare genetic diseases (including muscular dystrophy and chronic granulomatous disease) as part of McLeod syndrome. This X-chromosomal recessive phenotype is characterized by a complete absence of XK cell surface proteins, and therefore indicates decreased expression of all red blood cell Kell antigens. For transfusion purposes, these patients are considered completely incompatible with all normal and K0/K0 donors.
- RP associated with hypogonadism and developmental delay with an autosomal recessive pattern of inheritance, apparently with Lawrence-Luna-Bardet-Biedl
Other conditions include neurosyphilis, toxoplasmosis (Emedicine "Retinitis Pigmentosa"), and Refsum syndrome.
Classification
The modern classification of the disease depends on the mechanism of hereditary transmission of the genetic disorder:
- autosomal dominant inheritance
- autosomal recessive inheritance
- X-linked inheritance mechanism
- caused by mitochondrial DNA mutations
Based on the nature of the disease and the affected areas, typical and atypical forms are distinguished.
There are also classifications according to the presence or absence of concomitant abnormalities and according to the age of manifestation of the pathology (congenital, juvenile).
Koat's retinitis is a pathological development of the blood vessels of the eye. The disease is accompanied by bleeding from the affected vessels in the posterior wall of the retina, and against the background of increased cholesterol levels, it eventually causes retinal detachment.
Rheumatic retinovasculitis - appears as a white formation located at the location of the vessels. With late diagnosis, it results in a stable loss of visual acuity and the development of secondary perivascular fibrosis.
Toxoplasmic retinitis is manifested by the formation of scarring and atrophic changes without an inflammatory process. Accompanied by clouding of the vitreous body.
Syphilitic choriopetinitis - causes swelling of the retina and nerve, hemorrhage and opacification of the vitreous body.
Cytomegalovirus retinitis - affects HIV-infected people, is infectious in nature, and is complicated by retinal detachment.
Viral retinitis - caused by influenza, adenovirus and herpes viruses, manifests itself in clouding of the retina. The pathology is eliminated on its own, and by restoring the ability to clearly perceive objects, a relapse is possible.
Septic retinitis is accompanied by damage to the retinal vessels. Caused by pyogenic bacteria in the patient's body. The progression of the anomaly causes swelling of the eye nerve, vitreous opacification and panophthalmitis.
Typical
Visual acuity (central and peripheral) with this form remains normal for a long time. However, at this time the rod system is damaged and pockets of pigment accumulation appear.
Atypical
Atypical retinitis, depending on the affected areas of the retina and the nature of the progression of the pathology, is divided into:
- central
- non-pigmented
- white-spotted
- pseudopigmentous
- generalized hereditary retinal dystrophy associated with systemic diseases and metabolic disorders.
Symptoms
Retinal pigmentary degeneration manifests itself in adolescence; the dominant type can manifest itself at a more mature age.
The first symptom is a long adaptation of vision to poor lighting.
Further development of the disease causes the following symptoms:
- Periodic decrease in visual acuity.
- Disorientation in space and darkness.
- Difficult adaptation when moving from light to dark and vice versa.
- Good vision in daylight and blindness in the dark.
- Narrowing of peripheral vision.
- Distortion of the outlines of objects.
- Formation of blind spots in the field of view.
- Tunnel vision (narrowing of the visual field).
- Immunity to shades of blue.
Attention! Gene mutations affect the condition of the blood vessels of the eyes, this causes the destruction of cones, clouding of the lens, thinning of the sclera, and retinal edema.
Clinical triad
During ophthalmoscopy, a triad of symptoms is observed:
- bone bodies are typical foci of retinal hyperpigmentation;
- narrowing and atrophic changes in the fundus arterioles;
- pallor of the optic disc (waxy appearance)
Diagnostics
To make a diagnosis, the doctor needs to know which gene led to the onset of the disease and the distribution of such mutation in the family tree.
The diagnosis of retinitis pigmentosa is established in the presence of specific defects in the vitreous body.
Important! Children can be diagnosed with this after 6 years of age, when the child’s visual organs are fully formed.
Pathology research methods:
- taking anamnesis of close relatives
- examination of the fundus with dilated pupils and multiple magnification
- color perception testing
- assessment of the condition of eye vessels using fluorescein angiography
- determining the degree of adaptation to light and darkness using electroretinography
- Electrooculography – determination of the amplitude of vision in both eyes
- CT scan
- molecular genetic tests
Known cases
- Derrick Morgan, Jamaican ska, rocksteady and reggae musician.
- Neil Fachie, British Paralympic cyclist. [29]
- Lindy Howe, Australian tandem cyclist and swimmer [30]
- John Wellner, American actor [31]
- Steve Wynn, American business magnate and casino developer in Las Vegas [32]
- Rachel Leahcar, Australian singer
- Willie Brown, former mayor of San Francisco
- Steve Lonegan, Mayor of Bogota, New Jersey, Republican candidate for US Senate
- Amanda Swofford, American model[33]
- Richard Bernstein, Michigan Supreme Court Justice.
- Ian Treherne, British photographer.
- Matthew Benton, cabaret superstar and owner/managing director/artistic director of the Matthew Benton Dancers.
- Molly Burke, motivational speaker, blogger.
Treatment and prognosis
Retinitis pigmentosa is a little-studied disease that cannot be treated, but there is a chance of not aggravating the condition and preserving vision.
Lack of treatment can cause the development of concomitant eye diseases (cataracts, keratoconus, etc.).
Measures to maintain eye health:
- Preparations for maintaining vision and protecting the eyes - lutein, zeaxanthin, lycopene, blueberry extract.
- Strengthening the vascular wall (rutin).
- Low cholesterol diet.
- Diuretics for retinal edema.
- Vasodilators for better oxygen supply to the organs of vision.
Modern science is working to develop new treatment methods:
- Transplantation of cells responsible for pigmentation. Defective cells are replaced with healthy ones, as a result of which genes and chromosomes become in the correct sequence.
- Retinal replacement helps to significantly improve the condition, but does not completely cure the disease.
- Implantation – transplantation of donor stem cells and tissues (a method under active development).
In general, the prognosis is unfavorable; supportive treatment can delay the onset of blindness by 5-10 years.
Useful video
How does a person with retinitis pigmentosa see: